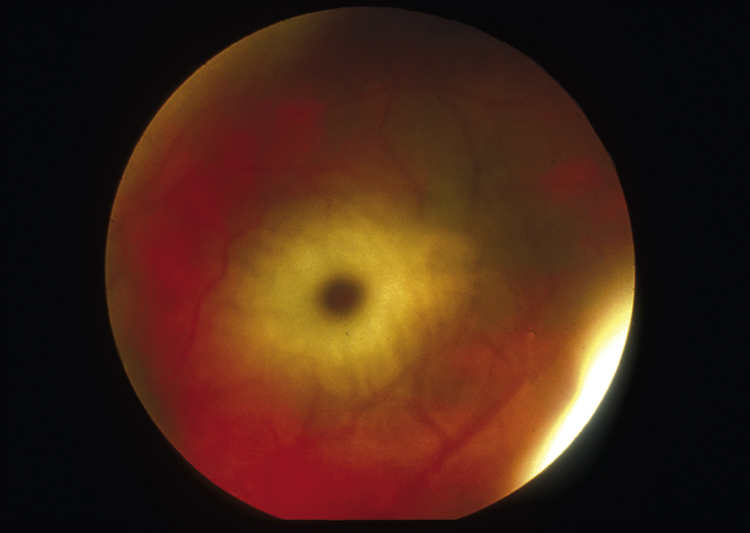
Tay-Sachs disease /tā″saks″/ [Warren Tay, English ophthalmologist, 1843–1927; Bernard Sachs, American neurologist, 1858–1944] , an inherited, neurodegenerative disorder of lipid metabolism caused by a deficiency of the enzyme hexosaminidase A, which results in the accumulation of sphingolipids in the brain. The condition, which is transmitted as an autosomal-recessive trait, occurs predominantly in families of Eastern European Jewish origin, specifically the Ashkenazic Jews. It is characterized by progressive cognitive impairment and physical delays and early death. Symptoms first appear by 6 months of age, after which no new skills are learned and there is progressive loss of those skills already acquired. Convulsions and atrophy of the optic nerve head occur after 1 year, followed by blindness, with a cherry-red spot on each retina; spasticity; dementia; and paralysis. Most children die between 2 and 4 years of age. There is no specific therapy for the condition, and intervention is purely symptomatic and supportive. The disease can be diagnosed in utero through amniocentesis. Also called amaurotic familial idiocy, gangliosidosis type I, infantile cerebral sphingolipidosis, Sachs’ disease. See also Sandhoff’s disease.