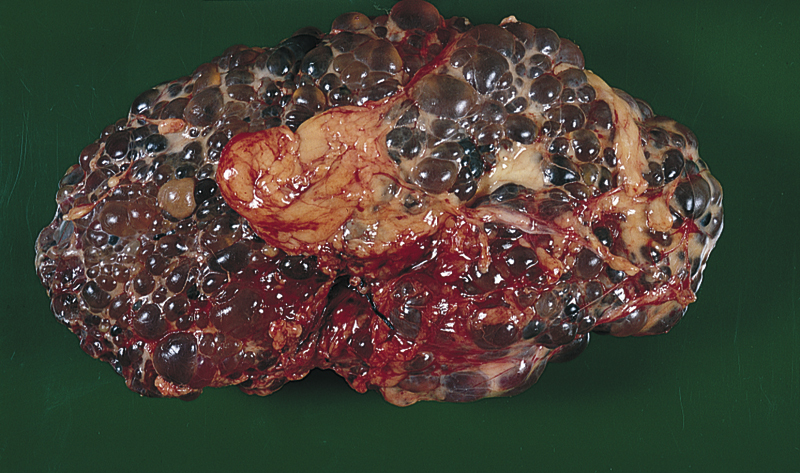
polycystic kidney disease (PKD), an abnormal condition in which the kidneys are enlarged and contain many cysts. There are two unrelated hereditary diseases in which there is massive enlargement of the kidney with cyst formation: Autosomal dominant polycystic kidney disease (ADPKD), formerly called adult polycystic kidney disease, is the most common type of cystic disease of the kidneys. It is usually manifested during the third decade of life. Renal failure may appear by the fifth decade, with terminal failure occurring in the next 10 years, although in some cases it never appears. Although there is rarely any liver dysfunction accompanying this disorder, cyst formation in the liver does occur. Autosomal recessive polycystic kidney disease (ARPKD), formerly called childhood polycystic kidney disease, is diagnosed at birth or in the first 10 years of life and is much less common than the autosomal dominant form. Both the kidney and the liver are involved, causing renal failure and liver failure with portal hypertension. Characteristic symptoms early in the process include pain, hematuria, urinary tract infection, kidney stones, and obstructive uropathy with anuria. ▪ INTERVENTIONS: Treatment of both types of polycystic kidney disease is largely symptomatic. Renal dialysis and kidney transplantation during end-stage renal disease can prolong life but offer no cure. Families with histories of polycystic kidney disease benefit from genetic counseling and may need help in coping with the prospect of future offspring affected with the disease.