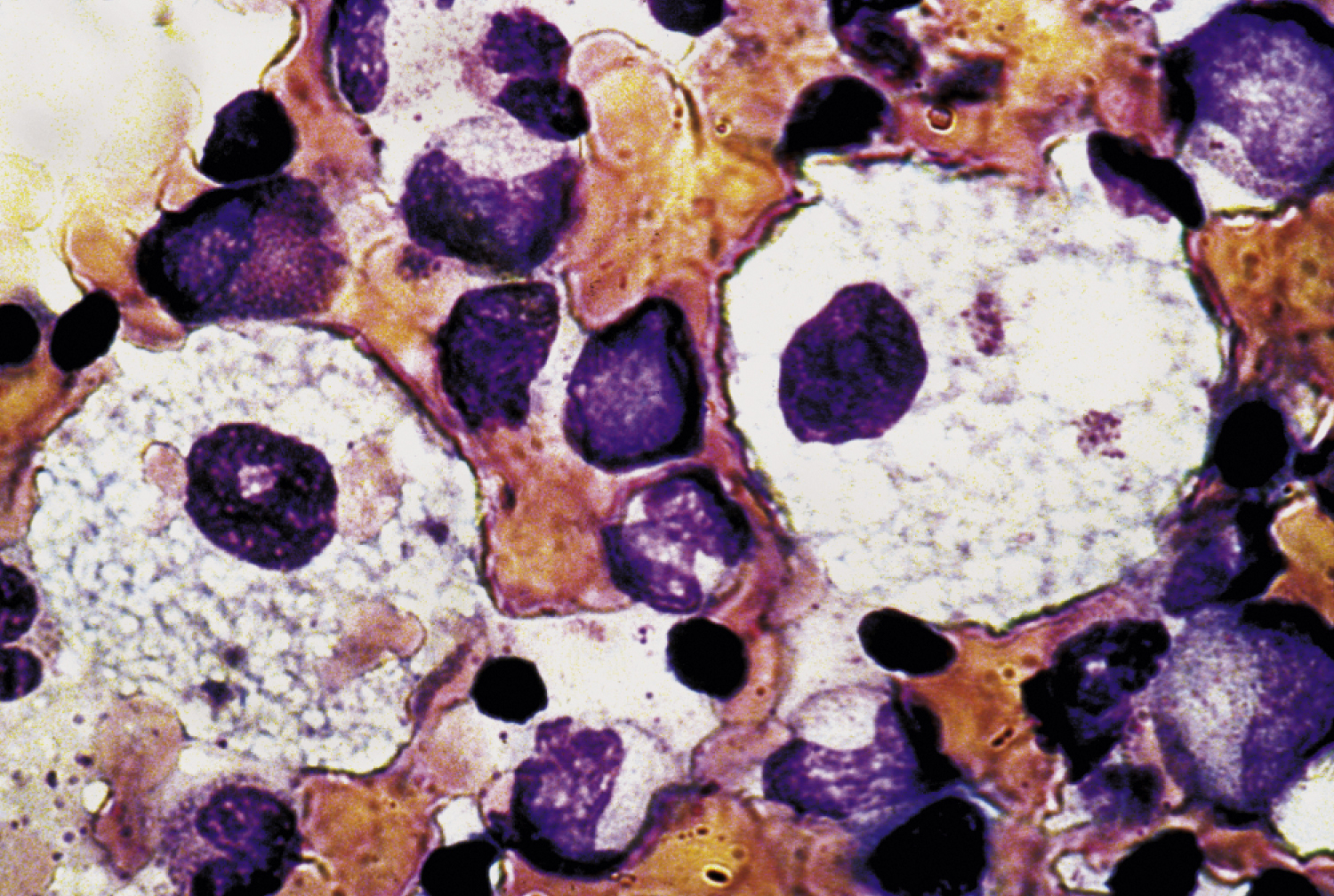
Niemann-Pick disease /nē″mon pik″/ [Albert Niemann, German pediatrician, 1880–1921; Ludwig Pick, German pediatrician, 1868–1935] , an inherited group of disorders of lipid metabolism in which there are accumulations of sphingomyelin in the bone marrow, spleen, and lymph nodes. There are several types of Niemann-Pick disease: type A (NPA) and type B (NPB), also called acid sphingomyelinase deficiency (ASMD); type C (NPC); and type D (NPD). NPA is seen in all races and ethnicities. In the United States and Canada there is a higher incidence of NPA and NPB among the Ashkenazi Jewish population. Type C is most common in Puerto Rican people of Spanish descent. Type D is seen in French-Canadian people in Nova Scotia. See also sphingomyelin lipidosis. ▪ OBSERVATIONS: Symptoms vary, depending on the type of disease. Generally, the disease is characterized by enlargement of liver and spleen, anemia, lymphadenopathy, and progressive mental and physical deterioration. Type A symptoms appear in infancy and are manifested by poor feeding, poor motor control, a cherry-red spot in the eye, and abdominal swelling. Type B symptoms are usually milder, occurring in late childhood or adolescence. Type C usually occurs in late adolescence or early adulthood. Blood and bone marrow tests are done to diagnose types A and B; a skin biopsy is the usual test to diagnose types C and D. ▪ INTERVENTIONS: There is no effective treatment for type A, and children with the disease usually die within a few years of the onset of symptoms. There are new treatments for types B and C. No specific treatment exists for type D. A well-balanced diet that is low in cholesterol is recommended. ▪ PATIENT CARE CONSIDERATIONS: DNA tests can be done to diagnose carriers of types A and B. In addition to the coordinated support of the health care team, support groups are helpful in assisting parents and children to cope with disease progression.