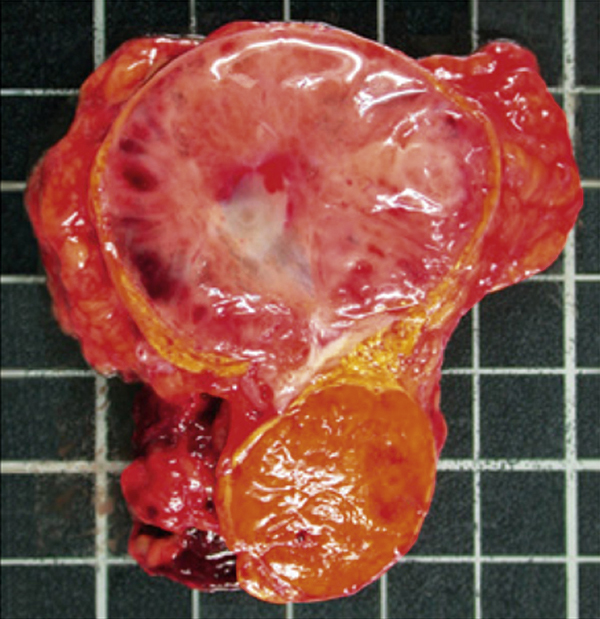
pheochromocytoma /fē′ōkrō′mōsītō″mə/ pl. pheochromocytomas, pheochromocytomata [Gk, phaios, dark, chroma, color, kytos, cell, oma, tumor] , a vascular tumor of chromaffin tissue of the adrenal medulla or sympathetic paraganglia, characterized by hypersecretion of epinephrine and norepinephrine, causing persistent or intermittent hypertension. Typical signs include headache, flushing, palpitation, sweating, nervousness, hyperglycemia, nausea, vomiting, and syncope. Weight loss, myocarditis, cardiac arrhythmia, and heart failure may occur. The tumor occurs most commonly at 40 to 60 years of age, and only a small percentage of the lesions are malignant. The cause is unknown. The diagnosis may be established by laboratory assays showing increased catecholamines and their metabolites in urine and by pressor tests; intravenously injected histamine causes a sharp increase in blood pressure, and the administration of phentolamine produces a marked decrease. Surgical excision is the usual treatment; patients with nonresectable tumors may be treated with adrenergic blocking agents or with methyl tyrosine, a drug that reduces norepinephrine production. Also spelled phaeochromocytoma. ▪ OBSERVATIONS: The most prominent sign is severe sustained or episodic hypertension. This is accompanied by a classic symptom triad of severe, pounding paroxysmal headache, palpitations, and profuse sweating. Visual disturbances, dilated pupils, lower extremity paresthesia, nausea and vomiting, dizziness, tremors, and tachycardia are also seen during paroxysm. An elevated plasma concentration of normetanephrine or metanephrine from a 24-hour urine collection is a 100% sensitive test for pheochromocytoma. A clonidine suppression test reveals persistent elevations in plasma norepinephrine. CT scans and MRIs are useful for tumor location. Complications that typically result from nontreatment or advanced disease include uncontrolled hypertension, diabetes, cardiomyopathy, cardiac arrhythmias, and heart failure. ▪ INTERVENTIONS: The primary treatment for pheochromocytoma is surgical removal of the tumor via laparoscopic adrenalectomy. Individuals are stabilized, starting about 2 weeks preoperatively, with a combination of sympathetic blocking agents and liberal salt and fluid intake. Preoperative palpation of the abdomen is contraindicated because it could cause sudden release of catecholamine and severe hypertension. Any persistent hypertension or postoperative hypertension after surgery is managed with conventional antihypertensive drug therapy. Individuals with nonresectable tumors are treated with alpha blockers or methyl tyrosine. ▪ PATIENT CARE CONSIDERATIONS: Careful monitoring of blood pressure is crucial in preoperative and postoperative periods to detect abrupt and severe fluctuations. Cardiac monitoring is done to detect cardiac complications, such as arrhythmia. Rest, nourishment, hydration, and emotional support are also needed. Nursing plays a crucial role in screening and case finding. Screening should be done for anyone who exhibits signs of malignant or paradoxical hypertension or displays a poor response to antihypertensive drug therapy. Individuals with neurofibromatosis are also at increased risk for pheochromocytoma.