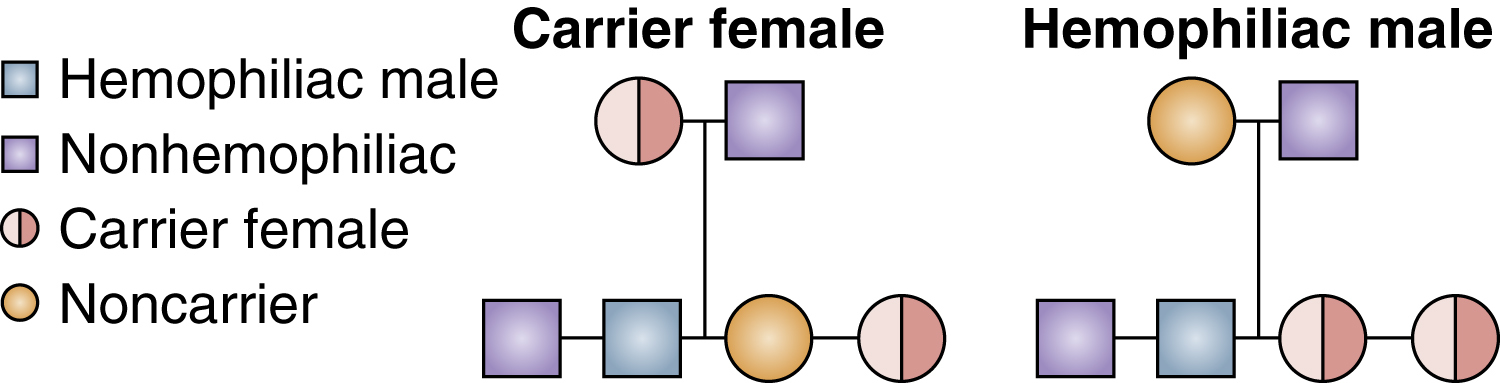
hemophilia /hē′mōfē″lyə, hem′-/ [Gk, haima + philein, to love] , a group of hereditary bleeding disorders characterized by a deficiency of one of the factors necessary for coagulation of the blood. The two most common forms of the disorder are hemophilia A and hemophilia B. Hemophilia A (classic hemophilia) is the result of a deficiency or absence of coagulation factor VIII. Hemophilia B (Christmas disease) results from a deficiency of factor IX. Hemophilia C (Rosenthal disease) is a factor XI deficiency. The clinical severity of the disorder varies with the extent of the deficiency. Anatomical bleeding—bleeding into joints, muscles, and soft tissue—is typical of hemophilia. Greater than usual loss of blood during dental procedures, epistaxis, hematoma, and hemarthrosis are common problems in patients with hemophilia. Also spelled haemophilia. See also von Willebrand disease, specific blood factors. −hemophilic, adj., −hemophiliac, n. ▪ OBSERVATIONS: The primary presenting sign in hemophilia is excessive, poorly controlled bleeding. The rate of bleeding depends on the amount of factor activity and the severity of the injury that caused the bleeding. A factor activity level below 1% can cause spontaneous bleeding or severe bleeding from even minor trauma. When the factor activity level is more than 5%, bleeding is usually caused by trauma and is more easily controlled. The bleeding is anatomical involving subcutaneous and muscle tissue, or deep bleeding into joints and organ systems. Laboratory tests reveal a normal prothrombin time, a prolonged partial thromboplastin time, and a normal platelet function. Assays are used to determine the factor affected and the level of factor activity. A common cause of death is intracranial bleeds, which occur in about 10% of hemophiliacs and are fatal 30% of the time. Other complications of repeat bleeds include joint and musculoskeletal deformities, pericardial tamponade, airway compression, and uncontrolled hemorrhage. ▪ INTERVENTIONS: Replacement of deficient factors with recombinant factor products or plasma-derived factor concentrates is the primary treatment. This may be used as prophylaxis or to stop bleeding episodes. Desmopressin acetate is used to stimulate factor VIII in mild hemophilia A. Aminocaproic acid is given in cases of persistent bleeding unresponsive to treatment. Pain is controlled with acetaminophen or codeine. Aspirin and NSAID use and intramuscular injections should be avoided because they may precipitate bleeding. Extreme care must be exercised when these individuals need surgery or dental work.